RAREFAN (RAyt/REpin Finder and ANalyzer) Manual
Using the online tool
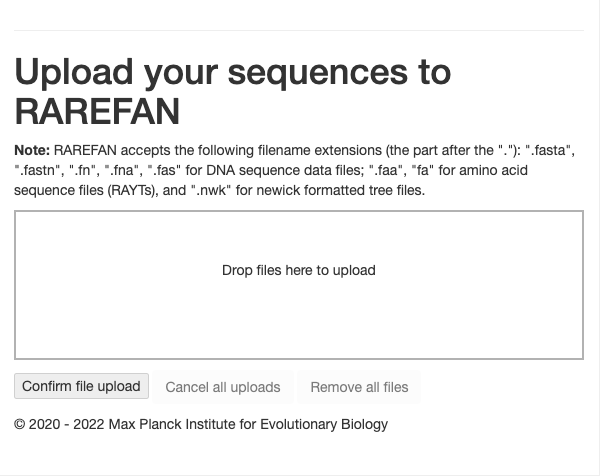
Here you can upload the genomes
that are to be analyzed for their REPIN content. The genomes should be in fasta format and only have the specified file extensions.
You can also upload RAYT query
sequences. These should be fasta files containing aminoacid sequences and should end in “.fa” or “.faa”. If these are not provided then you can select a RAYT
gene from P. fluorescens SBW25 (Group 3 RAYT (1)) or one from E. coli (Group 2 RAYT (1)) provided by the server on the next screen.
Finally, it is possible to upload
a phylogenetic tree in Newick format for your fasta sequences. For the webserver to be able to use the
provided tree the leaves of the tree have to match the sequence file names. If
the tree is not provided then a tree will be reconstructed using andi (2).
(1)
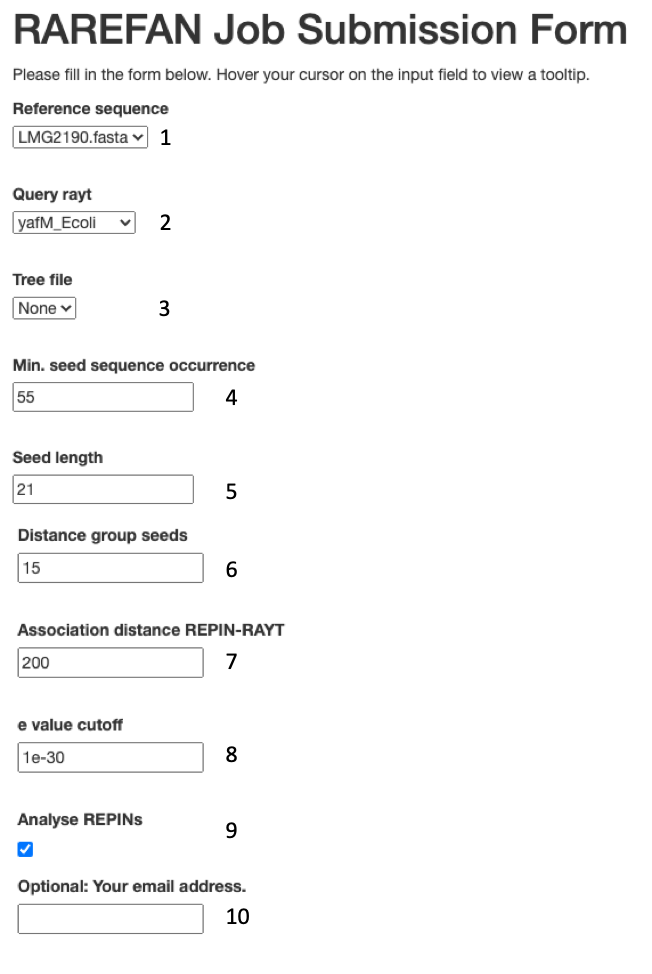
Please select a reference sequence from the
sequence files you submitted. The reference sequence will be used to identify a
maximum of eight seed sequences. The seed sequences have to occur at least n
times (specified in (4)) and are of length N (specified in (5)) and are the
basis for determining sequence groups. The way sequence groups are created is
described in detail in publication (3), in the section “Grouping
of highly abundant oligonucleotides in SBW25”. If sequence groups are part of
REPINs, then each sequence group can be used to define a REPIN type.
(2)
There are two very divergent RAYT groups (E.
coli group and P. fluorescens group).
If you do not know what kind of RAYT is present in your genome then you may
have to run RAREFAN twice, once with the SBW25 and once with the E. coli RAYT.
If you have supplied a RAYT protein yourself you should be able to select it
here. These sequences are then used as query proteins in a TBLASTN (4) search of the
provided genomes. All sequences that are identified below the e-value threshold
set in (8).
(3)
If you have provided a tree file in the previous
step then you can select it here.
(4)
This parameter is required for identifying
REPINs. Only sequences that occur more frequently than this value will be
considered when identifying REPINs.
(5)
The seed sequence length for identifying REPINs.
(6)
If two seed sequences occur at a distance of
less than this value, are considered to be part of the same sequence group.
Larger values will lead to smaller sequence groups and smaller values will lead
to larger sequence groups.
(7)
If a member of a REP sequence group is closer to
a given RAYT than specified by this number then the REP group is linked to the RAYT
gene (they will get the same color in the RAYT and REPIN plots). Smaller values
will lead to fewer REPIN-RAYT associations and larger values will lead to more
associations.
(8)
This e-value determines, which genes are
identified as RAYTs in the genome.
(9)
If this box is ticked then REPINs are
identified. REPINs are defined as sequences that consist of two seed sequences
in inverted orientation that are found at a distance of less than 130 bp. If
the box is unticked then the seed sequences (REPs) identified as described in (1)
are used for all further analyses. This can be useful if REPINs are asymmetric
(i.e. there is for example a deletion/insertion in either the 5’ or 3’ REP
sequence). This is for example the case for E. coli REPINs.
(10)
If you provide an email address here you will be
notified once the job is finished. This is particular useful when applying
RAREFAN to large datasets.

Once the job is submitted, it is
assigned a unique identifier. If you have not supplied an email address you
will either need to keep the site open or remember the unique ID to access the
website later. If you have closed the website but know the unique ID you can
access your data by calling this address http://rarefan.evolbio.mpg.de/results?run_id= and inserting your ID.

Once the job is
finished you can access your data either by directly plotting the data
(description of the plots below) (1); or by
browsing through the folder structure online (2);
or by downloading the data and viewing it on your hard disk (3). You can also rerun the data with a different
reference or different parameters (4).
File output
All output
data is located in a folder called out/.
The output
files in the folder out/ include the following:
|
Files in out/ |
Content |
|
tmptree.nwk |
A
phylogenetic tree of all genomes generated with andi
(http://github.com/evolbioinf/andi/) and clustDist (http://guanine.evolbio.mpg.de/problemsBook/node1.html). |
|
[reference].wfr |
A file containing
the frequencies of all 21bp long sequences found in the designated reference
genome. |
|
[reference].overrep |
Contains all 21bp long
sequences that occur more frequently than n (default 55) times in the reference
genome. |
|
yafM_relatives.fna |
A file containing
the nucleotide sequences of all RAYT relatives identified with
BLAST+ in all provided genomes. Contains only sequences that are
longer than 240 bp. |
|
maxREPIN_[0-5].txt |
If REPINs of
that group were identified in the strain and the REPIN option is ticked then
it contains the most frequent REPIN identified for each sequence type in each
strain. If the REPIN option is not ticked then the most common REP sequence
is shown. |
|
presAbs_[0-5].txt |
If the
REPINs option is ticked during submission then it contains information on
RAYT and REPIN numbers are shown as long as the strain does contain at least one
copy of that REPIN type. If the REPIN
option is not ticked then only information on REP sequences are going to be available
even if it says REPINs in the file. Specifically,
information on the number of RAYTs, the number of REPINs, the master
sequence, the number of master sequences, the entire REP/REPIN population
size, the number of REPIN clusters that contain more than 10 sequences, all
REPINs in the population as well as all REPINs that differ to the master
sequences in at most three nucleotides. |
|
raytAln.phy |
Nucleotide
alignment of all RAYTs identified in each of the query genomes. |
|
raytAln.phy_phyml_tree.txt |
Phylogenetic
tree calculated with PHYML from the above RAYT alignment. |
|
rayt_[strain name].tab |
Contains
location information for each identified RAYT relative for each strain. The
files can be viewed with artemis (5). |
|
results.txt |
Contains for
each strain the frequency of the six identified 21bp long seeds. |
|
repin_rayt_association.txt |
Table
containing information on which RAYT (column 2) from which genome (column 1) is
associated with which REPIN group(s) (column 3). |
|
repin_rayt_association_byREPIN.txt |
Same
information as above but in a different format. |
|
repin_rayt_association.txt.fas |
Nucleotide
sequences of each RAYT gene from each of the genomes in FASTA format. |
There is a subfolder
called groupSeedSequences/. All 21bp long sequences
in the genome that occur more frequently than 55 times are sorted into 6
sequence groups. These sequence groups are stored in the following files:
|
Files in out/groupSeedSequences/ |
Content |
|
Group_[reference]_[0-5].out |
Contains all
seed sequences that occur more than 55 [default] times in the genome that
were sorted into the specific group. As well as the frequency of those
sequences in the reference genome. The most common sequence in each group is
used as a seed sequence to determine REPIN populations across all submitted sequence
files. |
|
Group_[reference]_[0-5].out.fas |
The same
information as above but in FASTA format. |
|
[reference]_words.tab |
Contains the
locations of all overrepresented 21bp long sequences in the reference genome.
This file can be viewed in artemis
(https://www.sanger.ac.uk/tool/artemis/) together with the reference genome
file. |
For each genome there are six output folders (ending in _0
to _5), corresponding to each of the six sequence groups (only if there are at
least six sequence groups that occur more frequently than the seed sequence
frequency threshold).
Each folder contains
the following files:
|
Files in out/[genome]_[0-5]/ |
Content |
|
[genome]_[0-5].dd |
Degree
distribution of the REPIN network, where each REPIN is a node. A REPIN is
connected to another REPIN if they differ in exactly one position. The degree
distribution is a histogram of the number of connections of all the
nodes. |
|
[genome]_[0-5].hist |
For the
largest sequence cluster determined by mcl (6) that consists of REPINs (two REPs in inverted orientation)
this file contains the number of REPINs in each sequence class. Sequence
class 0 is the master sequence. By definition the most common REPIN in the
sequence population. Sequence class 1 contains all REPINs differing in
exactly one position to the master sequence. Sequence class 2 contains REPINs
differing in 2 positions etc. |
|
[genome]_[0-5].mcl |
Contains the
clustering output by mcl. Each line contains the REPIN sequence IDs that
belong to the same cluster. Lines are sorted by cluster size. REPIN/REP
sequences are clustered on sequence similarity. |
|
[genome]_[0-5].mw |
Contains the
most common 21bp long sequence of this group and its frequency in the genome,
which is the basis for identifying all related REP sequences and from those
the REPINs formed by these REP sequences. |
|
[genome]_[0-5].nodes |
The identity
and frequency of all REPINs and REP sequences. |
|
[genome]_[0-5]_largestCluster.nodes |
The identity
and frequency of all REPINs or REP sequences in the largest REPIN/REP
sequence cluster. |
|
[genome]_[0-5].ss |
Contains
REPINs and REP sequences as well as their positions in FASTA format. Position
information starts with the location in genome FASTA file (first sequence is
0...) followed by the start and end position of the entire REPIN/REP
sequence. |
|
[genome]_[0-5]_largestCluster.ss |
Same
information as above just for the largest mcl cluster. |
|
[genome]_[0-5]_[mcl cluster
number].ss |
The same
information as above except that it contains the sequences for a specific
cluster identified by mcl (see file *.mcl). |
|
[genome]_[0-5].ss.REP |
REP sequence
information in FASTA format. |
|
[genome]_[0-5].tab |
Location in
tab format. Can be used to display locations of REPs and REPINs in the genome
via artemis. |
|
[genome]_[0-5]_[mcl
cluster number].tab |
Contains the
location of REP/REPINs for each subcluster separately for viewing in artemis. |
|
[genome]_[0-5]_allSeed.nw |
Contains
network connections between nodes of all sequences. Can be used to view
network in for example R or cytoscape together with
the nodes file. |
|
_rayt_repin_prox.txt |
Contains
information on which REPIN/REP cluster is in the proximity (within 200bp) of any
of the RAYT genes identified in the genome. |
|
subfolder
[genome]_[0-5]/ |
Contains the
complete sequences (including the variable region) for all identified REPs
and REPINs. |
Data Plots
Here you can select the REPIN/RAYT type that is being plotted.
This is, for example, the data that is stored in the file presAbs_[number].txt.
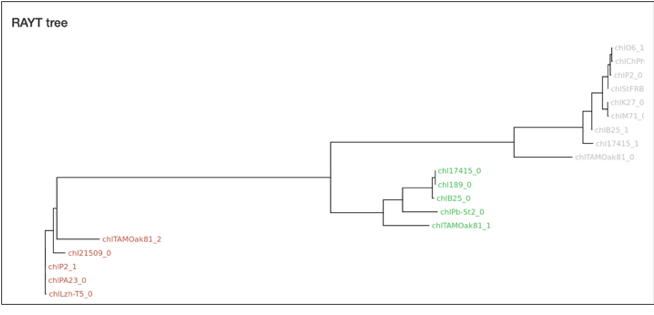
The first plot shows the
relationship between the RAYT genes and the REPIN type each RAYT gene is
associated with. The tree was generated from a multiple sequence alignment of
RAYT DNA sequences using the program MUSCLE (8). The tree itself is built using PHYML (9). The colors of the tip labels correspond to the
associated REPIN types. Colors are usually monophyletic due to a strong
association between RAYT and REPIN type.
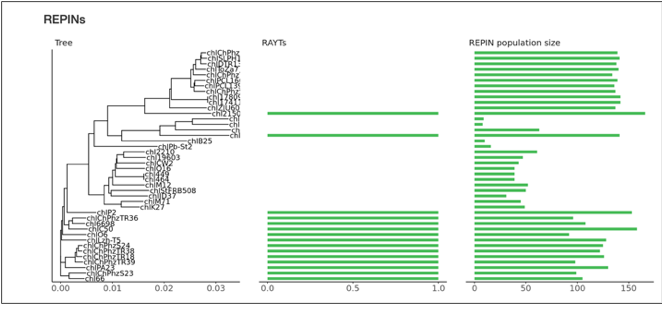
The second plot shows the number of RAYTs and REPINs
per genome. The tree on the left side of the figure shows the phylogeny of the
submitted genomes. The tree was built applying neighbor joining (7) to a distance matrix generated with the program “andi” applied to whole genomes (2). The next column shows the presence and absence of the
associated RAYT transposases. A RAYT transposase is considered associated when
a REPIN of the type is found within 200bp of the transposase. The REPIN
population size in each of the genomes is shown in the last column.
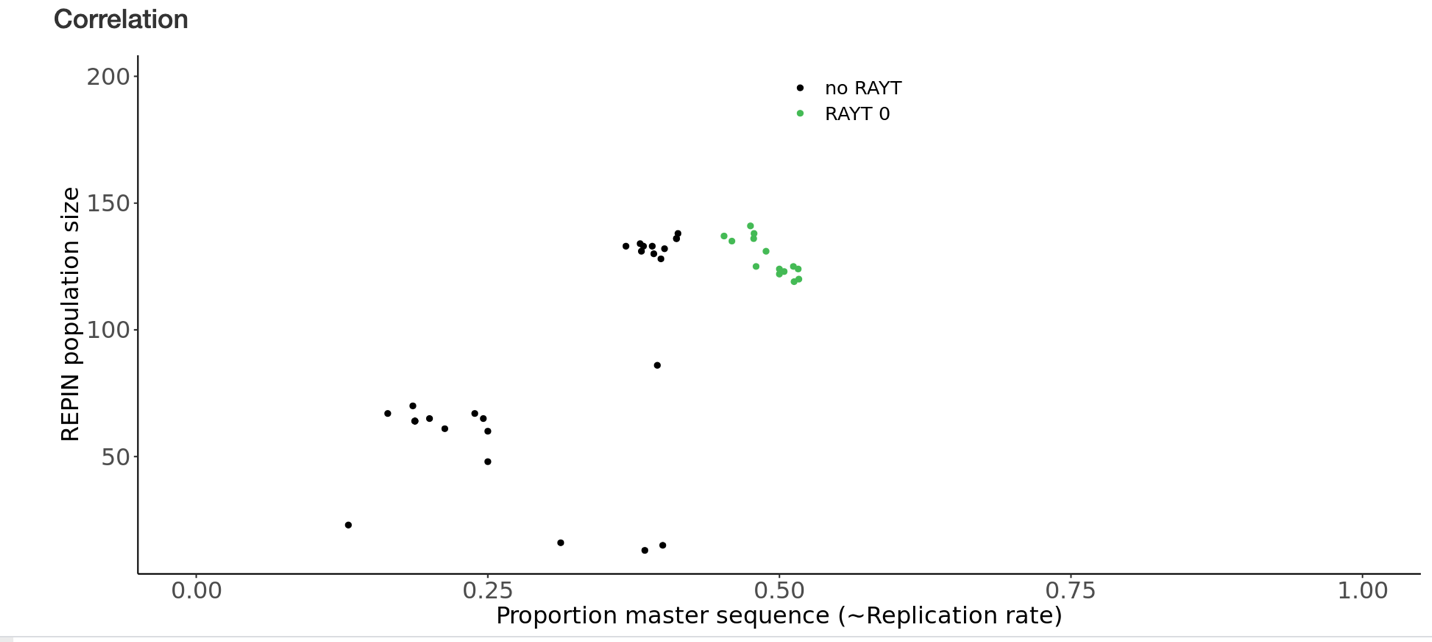
The proportion of master
sequences (indicates sequence conservation) in a REPIN population and the REPIN
population size. According to Quasispecies theory or mutation-selection balance
(10, 11), the higher proportion of master sequences (the most
common sequence in the population) correlates with higher duplication rates of
the sequence population. The closer populations are to the lower left of the
plot, the smaller and more decayed they are and the less likely they are to be
alive (i.e. actively replicating). Only populations that are colored are
associated with a RAYT transposase.
References
1. F. Bertels, J. Gallie, P. B. Rainey,
Identification and Characterization of Domesticated Bacterial Transposases. Genome
Biol. Evol. 9, 2110–2121 (2017).
2. B. Haubold, F. Klötzl, P. Pfaffelhuber,
andi: fast and accurate estimation of evolutionary distances between closely
related genomes. Bioinformatics 31, 1169–1175 (2015).
3. F. Bertels, P. B. Rainey, Within-Genome
Evolution of REPINs: a New Family of Miniature Mobile DNA in Bacteria. PLoS
Genet. 7, e1002132 (2011).
4. C. Camacho, et al., BLAST+:
architecture and applications. BMC Bioinformatics 10, 421–9
(2009).
5. K. Rutherford, et al., Artemis:
sequence visualization and annotation. Bioinformatics 16, 944–945
(2000).
6. A. J. Enright, S. Van Dongen, C. A. Ouzounis,
An efficient algorithm for large-scale detection of protein families. Nucleic
Acids Res. 30, 1575–1584 (2002).
7. N. Saitou, M. Nei, The neighbor-joining
method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol.
4, 406–425 (1987).
8. R. C. Edgar, MUSCLE: multiple sequence
alignment with high accuracy and high throughput. Nucleic Acids Res. 32,
1792–1797 (2004).
9. S. Guindon, et al., New algorithms
and methods to estimate maximum-likelihood phylogenies: assessing the
performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
10. F. Bertels, C. S. Gokhale, A. Traulsen,
Discovering Complete Quasispecies in Bacterial Genomes. Genetics 206,
2149–2157 (2017).
11. F. Bertels, P. B. Rainey, “REPINs are
facultative genomic symbionts of bacterial genomes” (2021).